BACK
TO ARISTOTLE? 1. ON URANOCHEMISTRY
B. Lukács
CRIP RMKI, H-1525 Bp. 114. Pf. 49.,
lukacs@rmki.kfki.hu
0. FOREWORD TO THE SERIES
Aristotle of Stageira
is the Prügelknabe of History of Science since the
French Enlightment, c. 1750. As you know, the Prügelknabe is cca. scapegoat; but in
a more direct way. Originally the Prügelknabe was a
substitute boy beaten if the young son of the Fürst
gave a wrong answer to the tutor.
For 250 years Aristotle is beaten
instead of (or: together with) the Catholic Church by Historians of Science (h.
of sc.). Any ancient Greek scientist other that Aristotle may have told
anything: historians of science are still enthusiastic telling that it was
originally a congenial idea. Platon may have
discussed the Soul which grows wings in ten thousand years and then flees above
the sky; h. of sc. tell what a nice allegory. His
nephew Speusippus may have written a morally terrible
letter to Philip of Macedon; h. of sc. do not discuss
this letter telling that it may not be genuine. Speusippus'
son, Eurymedon, may have accused Aristotle for asebeia (roughly: a mortal sin against religion), h. of sc.
elegantly ignore the event. Only Aristotle has no right to err.
No doubt, this aversion originated
from the close ties between Aristotle and the Catholic Church. Since St. Thomas
Aquinas, the teachings of the Philosopher were in the fundaments of Church
Science. For some centuries almost anything which he had taught was regarded as
Truth.
There are old stories, e.g. when a
faculty colleague of Albertus Magnus was confronted
with a fly. He was told to count the legs. His answer was: if the Philosopher's
text were not clear, I should accept that a fly has six legs. (The story is too
nice. The Philosopher's texts are not unequivocal about the numbers of legs of
the fly. Some years ago my colleague and Aristotle expert, K. Martinás tried with a hypothesis that Aristotle counted the
spider's legs and applied the result on the fly by analogy. It is possible: a
fly is not only smaller than a spider, but also its legs move much faster. So
it is easier to perform the counting on a spider. Remember that the number of
human chromosomes was also miscounted up to the 1950's as 24 pairs, which is
the correct number for our closest living relatives but not for us.)
St. Thomas Aquinas listed a very few
points where he (and so later the Church) did not agree with Aristotle; they
were mainly in connection with Man's Immortal Soul. Well, he could not use the
gifts of Divine Revelation (albeit Sir Thomas Browne cites an antique author of
Jewish religion in
So, when Enlightened Catholic French
freethinkers and anti-Roman Protestants find the Roman Church as common
adversary, they attack Aristotle as a symbolic figure. Platon
is harmless; he was simply a pagan.
Now, surely our present science is
better than the Philosopher's 2350 years ago. Also, the paradigm of the recent physics is different from the Aristotelian
one, except in Thermodynamics. (For
other sciences, and for social disciplines the
differences in paradigms are not so clear.) However, we are just learning that
Nature can be described in more than one way. And paradigms cannot be disproven in another paradigm.
Galileo believed he could prove the
Copernican system Now we know this would have been an
impossible task. In General Relativity you may choose any coordinate system whose origo you
consider at rest. In the Ptolemaic ~ Aristotelian description this is the center of Earth, and the (x,y,z) axes are fixed within Earth. With this choice
some "inertial forces" appear in the laboratory; the most spectacular
continuously changes the plane of the
If
you want to use astronomy and physics simultaneously, then you can do it
for first approximation more easily in the Copernican paradigm. Without physics
if you want to describe the motions of planets, it is simpler in a heliocentric
coordinate system, while for the lunar motion a geocentric system is more
convenient. But neither of them is "exact, so the true one".
But then, sometimes the Aristotelian
paradigm may be useful even in 2007 AD. My colleague, K. Martinás
succintly called my attention to this fact some
twenty years ago.
ABSTRACT
Uranochemistry
does not exist in post-Aristotelian paradigms (e.g. in Galilean-Newtonian one)
because Heaven does not exist there. Also, Aristotle did not work out uranochemistry because he believed no changes in Heaven.
Now I define uranochemistry as the most convenient
chemistry of meteorites & comets. Uranochemistry
obviously belongs to an Aristotelian viewpoint (Earth vs. Heaven); you may
decide if it is useful or not.
1. INTRODUCTION
We now think that, as a result of 4
centuries of evolution, our chemistry is quite good,
and this is of course true. However, the evolutionary path was not the only possible
one; and the specific evolution resulted in a chemistry,
which is still “too aqueous”. This is not a criticism; I do not claim that we
would be better off in alternative schemes, and definitely the massively
aqueous chemistry of Lavoisier and Arrhenius was a practical scheme to answer very practical
questions. I only tell that different evolutionary paths were possible too
which might have been more practical to answer other sets of questions (say:
extraterrestrial ones).
The great anti-Aristotelian fervour
starting in the time of Galileo & Newton has made any scheme in any science
impossible or at least very heavily unfavoured in
which the particular science would have location-dependent variants. Good:
What follows here will demonstrate
the possibility of an alternative way, whose idea comes from the
“solvent-system” way of defining acids & bases; only observe that there are
huge regions of World where there are even no solvents at all. I do not recommend the scheme of this study
at all; but maybe it is useful to look at situations from alternative
perspectives too.
Aristotle was a genius, 2350 years
ago. We have corrected almost all answers of his. But this in itself is not a
proof that his schemes would have been inherently wrong; and definitely the
corrections did not prove that he would have been stupid. I would be curious
how many of our nice answers will remain valid after 2 more millennia.
2. ACIDS, BASES AND NEUTRAL SOLVENTS
Acids and bases are very important
in post-alchemic chemistry, but acidity and basicity
are not absolute properties and even the definitions vary. For acids &
bases several different definitions exist which agree only in that in aqueous
chemistry hydrochloric or acetic acids are indeed acids, while sodium hydroxide
is a basis.
The story starts with Lavoisier, at the end of XVIIIth
century. This was the century finally substituting the Aristotelian (but rather
Theophrastean) alchemy with the modern chemistry. Lavoisier found that lots of oxidised substances form acids with water, for which sulphuric acid
is an obvious example. You burn sulphur; then you get SO2 or SO3,
and with water they form H2SO3 or H2SO4.
The term oxygen was coined from Greek (but not known by the ancients) meaning cca. “sourness-generating”.
Lavoisier
was not general enough, as we can see on the example of the hydrochloric acid, HCl. This was soon recognised by Davy, and later by Liebig. They told that the hydrogen was the key: but of course not all H, but H’s which can
easily be replaced by metallic atoms. Good; but why?
The answer came in 1884 from Arrhenius and Ostwald: chemicals
are of course dissolved in water, H2O. Now, a small part of H2O
is dissociated to H+ (or H3O+) and
This is still the chemistry below
university level. But Science goes forward, and 1923 was an annus
mirabilis for the theory of acids & bases: Brönsted
[2], Lowry [3] and Lewis [4] made new, more general definitions. The first two
defined acids as giving protons (so having a loose –H), while bases accept the
proton; Lewis defined backwards as bases giving electrons, and acids accepting
them. Surely, the Brönsted definition is more
generals than the Arrhenius one, but almost the same for water as solvent. Namely, take
hydrochloric acid and sodium
hydroxide basis, as reacting in water. HCl
“donates” its H; NaOH accepts it, but in such a way
that its –OH fuses with the “donated” –H. The result is HOH, water, and NaOH, salt.
So in water the Brönsted-Lowry definition
agrees with the older Arrhenius one. And in other solvents?
Usanovich,
Lux & Flood made even more general definitions,
which I ignore here. However Haldane [5], and later Firsoff [6] took
another way, to generalise the definitions for extraterrestrial situations.
This way is called the solvent-system
definition, and in a work about Uranochemistry I
should better take this way.
For a few subsequent paragraphs you
should forget that you are human born, raised and schooled on Earth.
Consider a situation when there are
no solvents at all, e.g. chemistry without liquid phase(s). Then it is not
natural to speak about acids. You may still, from the memory of liquids, call solid
or gaseous HCl as hydrocloridic
acid, but why? On the other hand,
hydrochloride HCl of course will have all the
absolute properties of the compound.
Now consider an exotic (for us)
environment where on a planetary surface there is ample liquid phase, but
predominantly HCl. In such an environment HCl would not look like an acid, because it would be simply
the general solvent of the environment. Such planets are rather improbable, but
only because of the cosmic element abundances: O is generally much more
abundant than Cl [6]. The explanation comes from
cosmology and nuclear physics and I would not go into the details here.
If you have a planetary surface
where liquid phases are accidental, various and negligible for quantities, you
can make liquid state chemistry, but still would not classify the compound according
to the behaviour in one solvent. Such a situation is expected for the most
planetary surfaces of the Galaxy, definitely if surface temperature &
pressure are not fortunate. Of course, autochtonous
life is not too probable on such planets, so aboriginal chemists are probably
absent.
Now consider a planetary surface
where the (p,T) range is
appropriate for liquid H2O.
For cosmologic reasons nuclei were building up by starting from p, d, n and a and not decaying from U. So H is the most
abundant atom and O, containing small, equal and even numbers of p and n is
quite abundant too even if much less abundant that H. Fig. 1 gives the raw data
for abundances; we will return to abundances later.
|
|
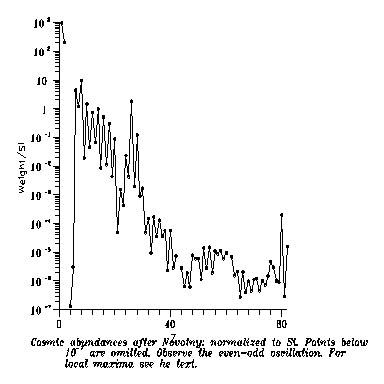
Fig. 1
As a direct consequence of Fig. 1, H2O
should be the most abundant compound
in stellar systems, although on the particular planet it may or may not be in
liquid phase. Still, if a planetary surface has a liquid abundant and dominant
enough to define a general solvent,
the biggest chance is that that solvent is water, i.e. liquid dihydrogenoxide, probably slightly sloppily called simply hydrogenoxide. (Observe that our sodium oxide is really not
NaO but Na2O. NaO
exists as Na2O2, but the situation is analogous to H2O2,
which is not hydrogen oxide, but hydrogen peroxide.)
So consider a hypothetical chemist
born and schooled on a watery surface. Surely he, instead of performing
difficult experiments with gases or very slow ones with solids, will start with
reactions in liquid phase, so he will try to solve his reagents in the abundant
hydrogenoxide, which he will call in a pre-chemical
name coming up from Palaeolithe, as water.
Dihydrogenoxide
easily dissociates to a positive, so electron-deficient ion -H+, and
a negative one, with electron surplus, hydroxile, -OH-.
So while the overwhelming majority of this “water” will be in dihydrogenoxide molecules, a small part will be in hydrogen
and hydroxile ions.
If our hypothetical chemist from wet
watery surface puts some sodium chloride, NaCl into
the “water” (surely, he will call NaCl on some
archaic name as sea salt or common salt or kitchen salt or such), the strongly
dipolar hydrogenoxide molecules will surround the
strongly dipolar sodium chloride molecules and “solve the salt”. The exact
process can be described in physics, and all good physicists of the Galaxy
would describe it very similarly. However for chemists from planetary surfaces
abundant in liquid hydrogenoxide the report would
generally be: “I had put salt into water, and a part of it went into solution”.
Indeed, physics tells us a somewhat
nontrivial prediction. If we put so much NaCl into H2O
that some NaCl “cannot dissolve” i.e. remains
together as molecules, then a tiny amount of two other compounds is created
too. Namely, there will be present the following neutral molecules and charged
ions:
H2O, NaCl, H+,
The explanation is
easy: first the 4 ions are created by dissociation, but then there is combinatoric possibility for the last two compounds to be
formed. If there is too much Na and Cl, a few HCl and NaOH molecules can be
detected by measuring apparatuses, not only the ions.
Well, from Neolithe
upwards HCl and NaOH were
surely familiar for the ancestors of our hypothetical watery chemist; of course
for millennia not as pure materials but in strong impure solutions. The
traditional names may be quite diverse, but surely touching or tasting them
would give strong and quite unpleasant feelings; after some methodical
experimentation it can be observed that the two compounds cause 2 different
feelings, and the unpleasant feeling can be diminished by adding an appropriate
quantity of the diametrically opposite material. After some millennia a precise
chemist may call this neutralisation.
But what does really
happen?
Let us start from a “strong
solution” of NaOH. That means H2O (so
common that our hypothetical watery chemist omits its explicit mention) and NaOH, the physicist calculates the quantum mechanics of the
reactor (e.g. a ceramic pot) and there will be present 2 molecules and 4 ions:
H2O, NaOH, H+,
Surely adding more NaOH the situation would go into the wrong direction;
simple experiments confirm this.
However, adding HCl
seems optimal; adding the proper quantity the unpleasantness ceases, and the
solution becomes “quite neutral”. Putting our hand into the pot we do not feel
practically anything; tasting the solution it is salty, but not unpleasant. The
physicist, from any planet, will get that the reactor (the ceramic pot)
contains
H2O, NaCl, H+,
and with equal numbers of the hydrogen and hydroxile ions. The salty taste comes from the NaCl molecules + the equal numbers of Na+and
Cl- ions, NaCl
being the “common salt”.
OK, this is understood by any
chemist born and schooled on a watery planetary surfaces:
this is conform with his early experiences back to toddler age, and the
traditions of his society beck to tens of millennia. But can he imagine the
chemistry of another society, from a planetary surface where there is an
abundant and dominant solvent, which is,
however, not his familiar hydrogenoxide?
Surely, his everyday terms would
mislead him. One can tell that an absolute
chemistry would be needed, one can define a “surface-independent”
chemistry, but for that first our chemist must turn to physics, as quantum
mechanics. Now first I will very briefly discuss the probable abundant and
dominant solvents, and then thumb rules come for some fundaments of a surface-independent
chemistry.
Fig. 1 shows that H dominates all
the other atoms; neglecting the passive He, by orders of magnitude. So probably abundant solvents would contain H. Then a
“negative” atom is needed in addition: Fig.1 shows that the most abundant candidates
are C, N and O. In our prescientific terms the
resulting solvents are H2O, water; NH3, ammonia or hartshorn spirit; and CH4, methane or marsh-gas.
Methane is a poor solvent for simpler inorganic compounds, but ammonia is
generally not worse than water, although ammonia is generally not so abundant as water. For liquid ammonia a somewhat lower
temperature is needed.
Now come
the thumb rules for chemistries with abundant and dominant but otherwise
arbitrary solvent in solvent-system
language. Let us look first a watery example but in nonwatery
terminology.
If
the general solvent is H2O
then an acid is a compound easily liberating a –H
ion,
and
a basic is one easily giving -OH
Then the
neutralisation is a process in which the positive H
and the negative OH ions appear without the
excess of any of them, as it was in the pure solvent, so e.g.
Much H-OH + Na-OH + H-Cl
« Much H-OH + More H-OH + Na-Cl
In our semiscientific
language:
Caustic soda + Hydrochloric acid « Common salt
(the neutral solvent is generally not mentioned in the semiscientific speech).
It is rather simple then to
generalize this scheme. Let the general solvent be
A+
+ B-
Then an acid is A+-C-,
a basis is D+-B-, a “neutral salt” is D+-C-,
and the other compounds do not classify into any of the 4 important categories of neutralisation.
E.g. consider a planetary surface
where the dominant liquid is hartshorn, NH3.
Then for a locally born & schooled chemist,
If the solvent is NH3, then acid is which
easily gives –H+ ion, basis is which
easily gives –NH-2 (amine), and a demonstrative
neutralisation is:
Much H-NH2 + Na-NH2
+ H-OH « Much H-NH2 + More H-NH2 + Na-OH
The reaction would be named by the ammonia chemist as
Sodic lye + Icic acid «
Common/Kitchen salt
(the names will be explained later).
Interestingly enough in laboratories we use this reaction to get really pure NaOH,
but with watery terminology as:
Sodium amide + Water « Caustic soda;
For more
information about non-aqueous solvents see e.g. [7] and [8].
3. ABUNDANCES IN THE TERRESTRIAL
CRUST
Fig. 1 shows the
atomic abundances in the stellar neighbourhood of Sol, deduced mainly from stellar
spectra. For details see [6]. This distribution should and may be approximately
valid globally for the Solar System as well because of the common origin of Sol
and the remaining part of his system; but surely not for the body of Earth. For
example Earth is depleted in volatiles as H and He. Of course we do not have
good sampling about the total Earth.
Again, Earth has
gravitationally and thermally been differentiated, so we expect the heavy or
highly melting components deep, in the neighbourhood of the center,
while the cool crust is probably composed of light/easily melting components.
We do not know the abundances for the crust but do know them for the upper crust, where "upper" is
defined by the depth available in sampling. Selection processes for Earth and
the terrestrial upper crust very much deform the primary abundances: the
selection function is shown as Fig. 2. As we see, H is not the most abundant
element in the upper crust (albeit not rare); O or Si
are more abundant. As a good example for the
selection, in the cosmic average Mg is roughly an order of magnitude more
abundant than Al (they are produced in very similar nucleosynthetic
processes, but Mg is an even-even nucleus while Al is not); still, in the
terrestrial upper crust the relation is the opposite. This distortion of
abundances is caused by (gravitational & thermodynamic) differentiation, whither we return
later.
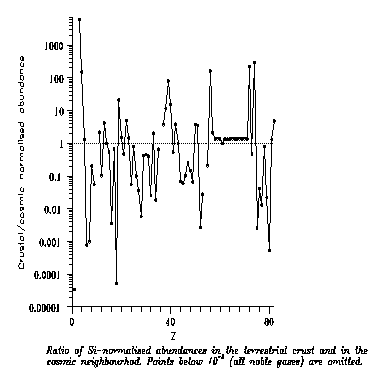
Fig. 2
4. ON OUR HEAVEN
Of course in the Solar
System abundances are changing from planetary body to planetary body. As for
the whole body this is the consequence of the primordial condensation process:
planets farther from Sol could collect more volatiles [9]. But surfaces differ
even more; the differentiation process had its pecularity
for each body. So there would be a specific chemistry for each surface. Of
course, there is the same physics on all the surfaces; however, if the name chemistry means more for us that simply applied physics, then the specific chemical rules change from surface to
surface. For example, with the possible exception of Mars and the Iovian satellite Europa, dihydrogenoxide is not the general solvent in the Solar
System outside Terra; and there is certainly no general solvent on Luna,
Mercury, Venus and the asteroids. And if you cannot naturally define the
general (neutral) solvent, then there is no natural definition or even meaning
behind the notions of acids, bases and neutralisation (except than some
terrestrial reminiscences).
In this work I call
everything "planet" in the Solar System which is a center of gravity, so of accumulation, in a neighbourhood
of non-negligible size. So Sol is a planet, Terra is a planet, Luna is a
planet, even asteroid Vesta is a planet. A planetary
surface is a place which significantly differs from the planetary body
(gravitational differentiation) and whence you can look upward to Heaven (so a
local center of spontaneous symmetry breaking from E(3) to SO(3) via gravitation is somewhere well inside the
planetary body). Heaven is practically the non-accumulated remainder of the
Solar System; negligible for mass but huge. And really it is Upwards, at least
from our actual coordinate system, adapted to the local symmetry breaking.
This Heaven contains
solar wind, comets, meteoroids, the smallest satellites and the smaller
asteroids; we believe that there is not real difference amongst the last 3
groups: meteorites are small asteroids accidentally meeting Terra and the
smallest moons of Jupiter and Saturn are just captured asteroids.
There are no planetary
surfaces in this Heaven; a very small asteroid has a surface in geometric sense
but otherwise not, the whole body is a homogeneous lump of matter. So there is
no liquid phase at all in Heaven.
On the other hand, we
have good sampling of the material composition of Heaven, thank to meteoritics and especially to the methodical work of the
National Institute for Polar Research in
5. ON THE ORIGIN OF
METEORITICS
In the recent decades
most people in meteoritics have originated as
geologists or in related topics. I think this is because you use very similar technics to analise a meteorite
and a terrestrial rock. Howevr geology had a quite
terrestrial origin.
In the Early Modern
Ages, say in XVIIth century, and especially after the
Thirty Year War,
Now, the simplest
important outward signal of suvereignity was of
course the army. Prince Louis Frederick Charles of Hohenloche-Oehringen
(360 sq. km, 25,000 pop.) had a few hussars, a hundred infantry, with officers,
a drummer and a trumpeter. However there definitely were smaller armies.
Dukedom Isenberg-Büdingen (12,000 pop., 60,000 fl.
income) had 20 soldiers, and County Limpurg-Styrum in
a time had an army of 1 colonel, 6 other officers and 2 privates. Of course,
this army was purely ceremonial, a mere declaration; but this is just my point.
(And observe that San Marino, 61 sq. km, pop. 17,000, had 180 soldiers in 1964;
state income 3.5 billion liras ~ 100,000 fl.)
Declarations of
independence were important because there were continuously some changes in the
Empire. Sometimes sovereign families died out, sometimes the Emperor split a
state between two sons (in
An item of prestige
was a University. It was not so cheap or simple as an
army, so the smallest states did not have their own universities; but there
were many universities in
A University is useful
in many ways. E.g. to the birthday of the Ruler the university staff can write
or read poems. But of course the Ruler disciplined the staff if needed. For
example Charles Theodore, Elector-Prince of the
While Geology is a honourable Science, geology, as evolved in Early Modern
Ages was somewhat influenced by the statuses of German universities. New
results are needed; but results not only new but also demonstrative. A new kind
of rock is good (a substantial part of Germany was a mining area), but a new
kind of rock which is purple with emerald green spots is even better; then the
Ruler, when visiting the University, will see how hardly the university works
and maybe even his Wife will remember the nice rock. The new mineral will be
immediately named; boring minerals will get the names of the discoverers, but
if something is purple with green spots, it will get at least the name of the
Countess.
Therefore emerging
Geology concentrated on Variety; it described thousands of minerals with
individual names.
Now, this great
variety goes back to terrestrial differentiation. Meteorites do not come from
Terra, and the majority never belonged to any planetary bodies. We can
recognise those which were formed originally in Mars, Luna and Vesta, the iron meteorites had been formed as cores of big
asteroids, demolished later, but almost all the others, and certainly all chondritic meteorites, have been formed in Heaven.
In Heaven there is no
General Solvent. So it is no point to define Acids & Bases (the definition would
be not only arbitrary, but pointless as well). Then there is no Neutralisation
either; and no liquid phase reactions. The Colourful Geology of the above
paragraphs could exist only on some major planets (Terra, Mars, Luna, Venus; we
do not see the surfaces of the giant planets); even big asteroid Vesta (whose crust we know quite well from diogenite, howardite and eucrite meteorites) could not have fed the universities of
Germany in XVIIIth century even for a couple of
decades. So Chemistry of Heaven (uranochemistry)
would not contain lots of complications present on the surface of Terra, so
could concentrate on other points. However first let us finish with the
planets, in order to see clearly what is the chemical
difference between Terra and Heaven and how we can be sure which
meteorite is of planetary origin and which is the product of Heaven.
6. PLANETARY
DIFFERENTIATIONS
There are good signals
showing that Sol, other planets and matter in Heaven have mainly common origin,
a galactic cloud starting to contract c. 4.55 billion years ago. External
contamination is probable but seems not too extensive.
The bigger asteroids
(surely upwards c. 100 km radius, maybe the borderline is somewhat below) are
spherical. So at the beginning even they were molten. While we still are not
certain, why and how, medium life time radioisotopes as Al26 or Pu244 may be behind. In liquid
state gravitational differentation is easy. So we can
visualize the evolutions of all rocky planets in, say, 6 consecutive stages,
as:
Stage 1: Everything is
molten. There is some gravitational differentiation (see Fig. 3 below), but
convection counteracts differentiation.
Stage 2: Cooling has
formed a thin solid crust; however the liquid below can regularly break it. In
terrestrial language we can speak about extensive lava flows; but this lava is
not too differentiated, it is near to the bulk distribution of the planet. (But
metallic Fe is accumulating in the center.) This
stage is preserved in the diogenites of/from Vesta, and maybe the lherzolites
and oldest komatiites of Terra.
Stage 3: The crust has
become thicker, and the undifferentiated Mg>Al lava has difficulties to come
up (gravity tries to keep heavier Mg-silicates below, being they of high
melting point, some of them freezes midway, &c.). So Al>Mg lava starts
to cover the Mg>Al surface. On Terra the really old basalts are rather komaiitic, the newer ones rather not.
Stage 4: The crust has
become more thicker. Generally volcanism is in
separate spots, through long shafts. The matter reaching the surface is
generally light and of low melting point; both conditions prefer (Al,Ca) and even more Na to (Mg,Fe). See: eucrite (Vesta), basalts (Venus, Terra, Luna, Mars).
Stage 5: Some up to
now unknown mechanism disprefers Al to Ca on Luna and
Mars. On Terra there is an opposite preference, but this may be biologic. See
Fig. 4 for extraterrestrial basalts and Fig. 5 which include terrestrials as
well.
Stage 6: Volcanism
stops. For some time there is still post-volcanic activity of gases but this
does not leave permanent traces. Maybe Mars is now in post-volcanic stage; and
on Terra we know local examples, e.g. Hargita is the
only mountain still showing post-volcanism in the whole
When volcanism stops,
the surface cannot anymore be rejuvenated. Composition freezes or slowly
corrodes, depending on the atmosphere.
|
|
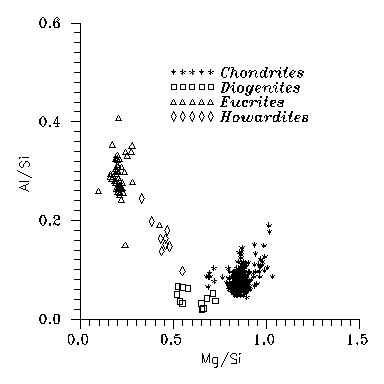
Fig. 3: Al vs. Mg abundances
for chondritic and Vestan
basaltic meteorites. Diogenites are still near to the
primordial composition.
|
|
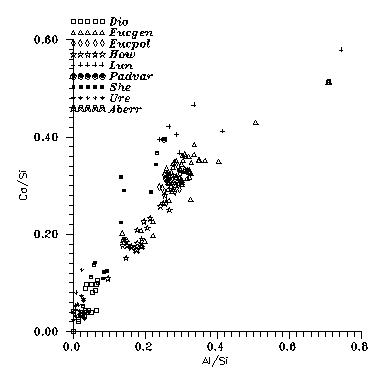
Fig. 4: Ca vs. Al contents for Vestan (Dio, Eucgen, Eucpol,
How), Lunar (Lun) and Martian (She) basaltic
meteorites + some others whose origin is still unclear. Observe that Lunar and
Martian basalts prefer Ca; for the others the Ca/Al ratio is cca. the chondritic
one which in turn is near to the cosmic abundance ratio.
|
|
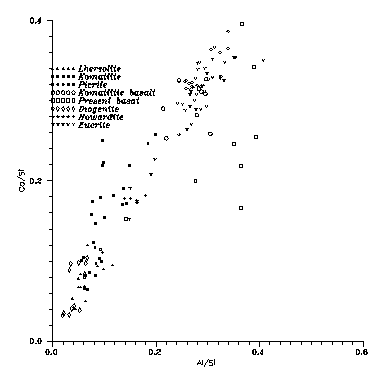
Fig. 5: Ca vs. Al contents
for Terran and Vestan
basalts. Observe that older Terran basalts (komatiites) deviate upwards
from the cosmic abundance ratio, while younger ones deviate downwards.
The undifferentiated
matter seems to be preserved in chondritic
meteorites. Fig. 3, at least for Al and Mg, show that they
have abundances similar to the cosmic one, and in fact for all elements going
easily to solid state this is more or less true. For eucrites,
howardites and diogenites
the textures show that the matter had been melted in the past, even if for diogenites the abundances would not disprove primordial
origin. We can recognise lunar & Martian basalts and lunar anorthosites as well, having lunar rocks at hand and probes
on Mars. Iron meteorites are surely remnants of cores of differentiated
asteroids demolished in the past. The crusts of these demolished asteroids are
certainly in the samples and experts have some guesses about them. All other
meteorites are heavenly.
7. ELEMENTS IN HEAVEN
While in principle all
elements of the periodic table which can be found in terrestrial natural
conditions do exist in Heaven too, some of them are extremely rare, so they are
not needed in first approximation to Uranochemistry.
For a guess see Fig. 1; but since elements can be suppressed in entering solid
phase, some corrections are needed too. We take 3 steps.
Fig. 6 is the first 4
rows of the periodic table, Si-normalised abundances
as in Fig. 1, but a different display. Noble gases, not participating in
chemical processes, are omitted. Fig. 7 is the same for the upper terrestrial
crust, so the analogue of Fig. 2. Since there are already more than 8 columns
in Row IV, each row is labelled with its most common element.
Now, Fig. 8 is the
same, but for the rare metorite class C1, maybe under
the minimal thermal impact in the past so most primordial, the meteorite Y-82162
from the NIPR collection. Elements beyond Row IV are rare in Heaven (see Fig.
1).
|
|
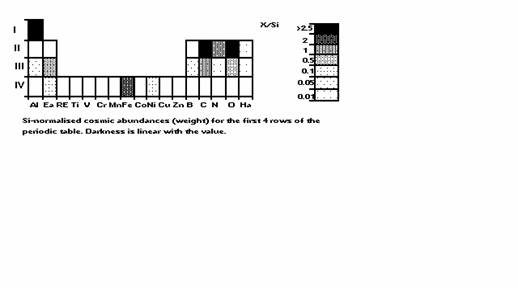
Fig. 6: Si-normalised
cosmic abundances for the first 4 rows of ther
periodic table.
|
|
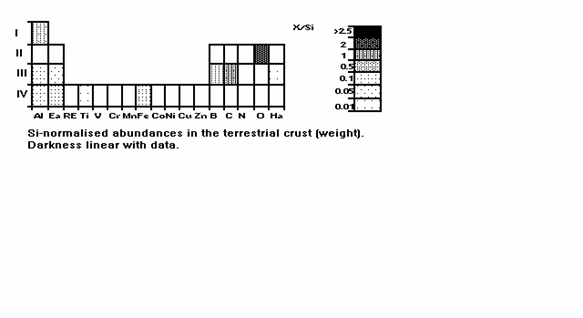
Fig. 7: As Fig. 6, for the
terrestrial upper crust.
|
|
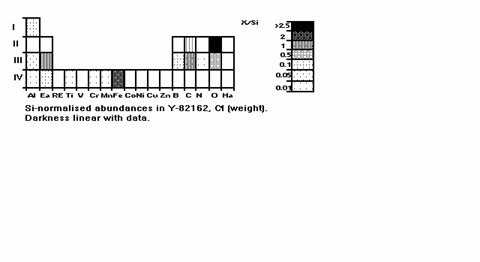
Fig. 8: As Figs. 6 & 7,
for the rather unaltered meteorite Y-82162.
Obviously in Chemistry
of Heaven all elements are important which are abundant there. However, for
minor components, we have another factor, standard
potential. Atoms of elements can yield an electron easily (e.g. a large
alkali atom its outermost electron above a "closed shell"), or not so
easily (a small alkali where even the outermost electron is near to the
nucleus, or an alkali earth one of the two outermost ones, leaving behind not a
closed shell, or such). Or they would rather not yield an electron but would
take one easily (a small halogen, which can make its outermost shell closed by
taking one), or still rather take than yield, but not so easily (a large
halogen, or a Column VI atom which will not have a closed shell even then, or
such). Standard potential is measured in Volts, well above +1 V for alkalis and
well below -1 V for halogens. It can be measured also for some molecules or
radicals if they do not disintegrate in the measurement.
Now, Fig. 9 shows the periodic system down again
to Row 4. If an element is rather rare and fairly inert, then it can be
overlooked in Uranochemistry and still the
description is good. Question marks appear for various reasons. Sometimes I did
not find the data. Sometimes the element simply does not produce ions in aqueous solution; and the standard, or electrode, or redox, potential was a par excellence aqueous quantity until
recently. (Now some data are available in liquid ammonia solvent, and they do
differ from those in water.) And carbon is the best example for uncertainty: while
it has no ions in aqueous solution, so the redox
potential cannot be directly measured, the solid state structure seriously
influences the redox behaviour of carbon. Amorphous
carbon reacts quite aggressively with oxygen, as we see it from the fact that
it can take away the oxygen from FeO. Diamond and
graphite burn also vehemently. But buckminsterfullerene, C60, is an electron acceptor, so “negative”, although it is
also pure carbon. So the electrode potential does not yield a final
understanding of chemistry in Heaven; still it gives some insight.
True, the values have been measured at 1 atm, and 25 centigrades, but in
first approximation they are determined by electron shell structure, so the
changes from lab to Heaven environment are probably minor.
|
|
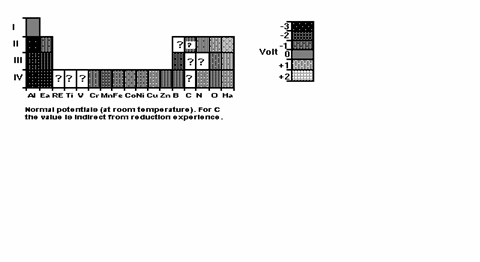
Fig. 9:
Now let us combine
Figs. 8 & 9 (or Fig. 8 could be replaced by one for all chondrites;
no big difference). I go from column to column; boldfaces stand for elements of first order importance in uranochemistry.
Column
1, alkalis. 3 elements occur with non-negligible abundances, but
K is much rarer than Na and the latter rather mimics K. So Uranochemistry
can work with two alkalis, a strong one, Na,
and a weaker one, H. Fig. 10, below,
will demonstrate the Na-H competition in chondrites,
but first we should finish the listing.
Column
2, alkali earths. Be is of negligible abundance. We remain with Mg and Ca.
Rare
earths. They are negligible.
Columns of Ti, V, Cr, Mn: Their abundances are
rather negligible.
The columns of the iron triad:
Only Fe is really abundant; Ni
generally occurs together with metallic Fe, and Co is rare anyway, so we remain
with Fe.
Columns of Cu and Mn: Their abundances are rather negligible.
Column 3: Only Al is abundant not to be ignored.
Column 4: Two elements
have non-negligible abundances, C
and Si.
Their roles are quite different (see later).
Column
5, of nitrogen. Interestingly enough, N is negligible in the
heavenly solid phases; I do not quite understand, why, but this is an
observational fact (causing difficulties to understand the Beginning of Life on
Terra, but that is another matter). P may or may not be omitted.
Column 6, of oxygen: Both
O and S are important and abundant.
Column 7, halogens: All
are of negligible abundances. However I believe
that for Cl
this is an artefact of terrestrial wet environment + aqueous analytic methods,
so I would suggest to keep it.
This is a scheme with
9 elements, rather similar for complexity to medieval alchemy of 5-10 elements.
Similarly to that, the 9 elements may represent also a few more (e.g. Fe
represents also Ni), but unsimilarly, we can be sure
that the neglection of the remaining 83 cannot really
influence the final results.
On another, more
Aristotelian, language we may call them as:
The weak alkali (H).
The strong alkali (Na).
The heavy earth (Mg).
The light earth (Ca).
The Jolly Joker (Fe).
The
The volatile (C).
The Rock Maker (Si).
The agressive non-metal (O).
The non-agressive
non-metal (S).
The halogene (Cl).
Fe is the Jolly Joker
because it may appear both in the metallic phase and in the non-metallic one,
with both valences 2 and 3, and both with O and with S. C is the volatile,
because its abundance changes very much with previous thermal history. Fig. 10
shows that Na and H substitute each other in Heaven; maybe the actual anticorrelation was caused by the competitions of the 3
alkali oxides Na2O, NaHO and H2O at primordial condensation. As for Rock Maker Si,
a new Chapter is appropriate.
|
|
Fig. 10: Na-H competition
for C2 and C3 meteorites.
8. ON PROCESSES IN HEAVEN
In all stone known meteorite the texture is determined by an SiO2 lattice. In older or more conservative
geology/chemistry books you can read that the silicates are salts of the silicic acid, H2SiO3.
Indeed, in terrestrial chemistry, with H2O as general solvent, there
are reactions suggesting such a view. They go mainly backward, but even then...
If you heat up meta silicic acid H2SiO3, it slowly yields water,
and amorphous quartz remains, as
H2SiO3
→ H2O + SiO2
So in aqueous chemistry quartz sand seems to be the anhydride of silicic acid. The statement is almost meaningless, however,
because it is almost impossible to solve quartz sand in water.
Still, insolubility is
formally not a counterargument. See e.g. aluminiumoxide,
Al2O3. It is insoluble in water, however a formal
reaction Al2O3 + H2O would (if could) give aluminiumhydroxide Al(OH)3.
It is still hardly soluble in water, but reacts with acids in a regular
neutralisation.
Now, the sodium silicate,
which is fairly soluble in water, has formally some relation to a silicic acid as it can be seen from an inverse reaction:
Na2SiO3
+ H2O + 2HCl → 2NaCl + H2SiO3
+H2O
However
surely the silicates of the terrestrial crust have not been created in an aqueous neutralisation reaction of a silicic acid and a basis as e.g.
H2SiO3
+ Mg(OH)2 + much water → MgSiO3 + more water
because there was never enough H2O
in the body of Terra to solve the necessary amount of quartz sand or silicic acid, and according to Terra's reconstructed
history there was no significant quantity of Mg(OH)2 in Terra.
Really, the reconstructed terrestrial history goes into a quite different
direction: silicates directly condensated from the protosolar nebula. There is no known meteorite which would
have ever evolved in a dominantly aqueous environment. While C1 and C2
meteorites are "wet" even in them the dihydrogenoxide
is not the dominant component.
There were never solvation processes in Heaven. On the other hand, at
Beginning of Times (at the Birth of the Solar System) there was condensation
from hot (or, far out, tepid) protosolar gas. Of
course we could tell that there is only one chemistry;
but then it could not include acids & bases whose classifications do depend
on the solvent; and if there was and is no solvent at all...?
Going back to SiO2, in some strange way, SiO2
plays a similar role in uranochemistry (but in solid phase)
as H2O does in terrestrial chemistry. Stony meteorites are dominantly
silicates. Then in meteorites you can observe textures of, say, MgSiO3 as well as of Mg2SiO4. However, they are not really valence
compounds. Rather, the first is SiO2*MgO, while the second is SiO2*2MgO.
The SiO2 can build up an infinite lattice
in 3 dimensions, each Si being surrounded tetrahedrally by 4 O's and so on. Now, there are free
places in the lattice where MeaOb oxides can enter. But
the inclusion of metallic oxides of course deforms the silicon lattice.
Now, SiO2 is an analogon of
H2O in two ways. The first, more direct way is to look at SiO2 of uranochemistry
as the crystallic water of aqueous chemistry. The
second way is to tell that in some sense meteoritic silica is the "neutral
carrier" of metallic oxides, carbon, iron sulfide
and metallic iron. Many times we can qualitatively describe the reaction even
without speaking about the silica. Of course then even pyroxene is not much
distinguished from olivine.
9. CLOSING REMARKS
I am not criticising
usual chemistry. It does work well. Only, under extremely non-terrestrial
situations it is not the simplest way. Without lots of dihydrogenoxide
the statements about solubility, the acid-basis dichotomy and such are either practically
meaningless, or misleading, and in both cases we should forcibly forget them.
Now, for a demonstration of the usefulness of
local chemistries consider a dominantly liquid ammonia environment. I do
not know if it occurs anywhere in the Solar System (if so, then outward [11]),
and it definitely does not occur in Heaven, but it may occur somewhere (and
there is a mysterious observation about Origin of Life, see later). Now, how to
try to describe Chemistry of this Alien World?
On an ammonia-washed
planet autochtones would not be disturbed about our
ideas about acids, bases and salts. There the reaction of their rather simple
compounds, known in impure states since Neolithic time
NaNH2 +
HOH = NaOH + NH3
would really be read as: common
lye can be neutralised with icic acid, and they get
common salt/kitchen salt + moisture is liberated. Why would they read it in
this way?
Well, their common
solvent is NH3=H-NH2. Their tissues
are full with NH3. It is quite neutral for them, it does not attack
their bodies, their biology is the product of billion
years of evolution in NH3. (Of course, they would not call their general
solvent “ammonia”. And even in their Middle Ages they
did not call it “hartshorn spirit” because it was not
a specific material. Maybe “moisture”, or “sap”.) But NaNH2, our sodium amide, is a
quite common aggressive compound, a lye. Why?
The explanation goes
in two parts. First, ammonia dissociates (in a small amount) to H+ and NH2-. Ammonia is
neutral, having the above radicals in equal amounts. But if we add NaNH2, the ion balance
ceases. The solution will have a great excess of negative ions. So our sodium
amide is there a strong caustic, sodium lye.
In addition, their
sodium lye must be quite common. Namely, sodium oxide is only some 1 % in chondrites. However, sodium silicates are light and are melt at relatively low temperatures, so late volcanism
brings up Na-rich basalts. This is pure physics (gravity + thermodynamics) so
it is independent of the solvent on the surface. Therefore sodium must be a
common enough element, and some of its compounds are solved fairly well (in ammonia,
of course). Also there are reports that even metallic Na can be solved somewhat
in ammonia (I guess, because of the alkalic
metal-type behaviour of the NH4 group, see ammonium amalgam),
although we cannot expect too much metallic Na in natural environment. Maybe
their soda lye was first produced also from natural Na2CO3.
Now, HOH in NH3 environment gives the
positive ion of the solvent but not the negative one. So it acts on the
discussed extraterrestrials just as our acids do on us. But HOH must be an
abundant compound there, because ice is an abundant component of many stellar
systems (according to calculations) due to the high abundance of the elements H
& O (see Fig. 1). However in their environment HOH is
solid, a rather light rock. So it is a rock which is acidic if you taste
it, icic acid.
Now, if soda lie is
abundant and common, NaOH, spontaneously coming from
fairly common soda
lye and the abundant acidic rock is neutral, so can be eaten. Also, it contains
the radicals Na- and -OH. I guess, for them both are necessary, but surely at
least Na. Na is important for all terrestrial creatures, a useful metallic ion,
so why not there? So "common salt" is a good name. If it has even
some taste for them (why not?), then it may also be "kitchen salt".
Anyway, it is very probable that their seas contain NaOH,
because Na comes from sodium silicate and OH from the acidic light rocks found
everywhere; and then Life started in the seas and for sophont
land animals NaOH is matter of life and death to maintain the
primordial chemistry. Originally this was doubted: Ref. [5] tells that NH3 does not solve the alkali
chlorides. But NaCl is an exception; although ammonia
solves it less than water does, still a concentration similar than in seawater
is possible.
This few paragraphs
were the draft of an essay about ammonia-chemistry produced without lots of
quantum mechanical calculations. I am certain enough to write it down; if
somebody wants to check the reaction in ammonia solvent, a moderately low
temperature lab is needed, with breathing masks. Without liquid phase the
reaction is standard even in our laboratories.
However now comes the Mystery of Life. Terrestrial life uses amino acids
as LEGO building blocks of proteins, some 20+ rather small molecules from which
huge proteins can be put together.
Obviously such
building blocks should be "bipolar", having two opposite characters
at the two ends. Then if you make two to react with one positive and one
negative end, the result will be a bigger molecule again with a positive and a
negative end, and the process can be repeated.
In aquaeous
chemistry the most natural choice for a bipolar reaction is neutralisation. Also,
for long chains C is needed, so the thing must be done in organic chemistry, where the compounds must have a -COOH end to easily loose -H (obviously from reasons
understood only via Quantum Mechanics; I never calculated this. Really, the
radical would be better to write as one O with a double bound out of the plane
of the paper, and the other as -O-H; and then it would be easier to imagine that -COOH yields merely an
-H; but this would be a Figure, not a text.) Good, so the simplest organic
compound which is acid on one side and basic on the other (so an oxyacid) is the glykolic acid:
HO-CH2-COOH
(the trivially simple HO-COOH not working in the proper
way for symmetry reasons).
You might expect that
two glykolic acid molecules would react as
HO-CH2-COOH + HO-CH2-COOH = HO-CH2-COO-CH2-COOH + H2O
but it is not so. Rather the long molecule
rearranges as:
HOOC-H2C-O-CH2-COOH
and that double acid is a cul-de-sac.
Terrestrial life has
found the way out by using amino acids instead of oxyacids,
the first member of the sequence being then glycine:
H2N-CH2-COOH
Now, two glicines react in a proper way as
H2N-CH2-COOH + H2N-CH2-COOH = H2N-CH2-CONH-CH2-COOH + H2O
giving another molecule with an
amid and a carboxyl end, so that is another LEGO. (-CONH-, which again should
be a picture, is the peptide bound, characterising the proteins.)
That is really nice;
but on one end of the building blocks of Life there is not the familiar hydroxile radical of aqueous chemistry, but the familiar
amino radical of ammonia chemistry.
The peptide bound of
aqueous organic chemistry seems to belong to ammonia chemistry! (Wait a
minute.)How is this? And why is this? I do not know. But I can make an easy
prediction for life in ammonia planets. There a very similar trick is expected
to work, starting with proper bipolar molecules of ammonia chemistry:
H2N-R-CONHH
If -R- is the simple -CH2-, then the molecule exists also in
our aquaeous organic chemistry, and is a building
block of oxytocine. Now, put together lots of such
ammoniac aminoacides. With repeated ammonia loss a
chain builds up
H2N-R1-CONH-R2-CONH-...CONH-Rn-1-CONH-Rn-CONHH
Now,
H2N-R1-CONH-R2-CONH-...CONH-Rn-1-CONH-Rn-COOH
is the corresponding protein of aqueous
chemistry, and if the molecular weight is ~106, the difference in
the very last radical is relatively almost nothing, so if one polypeptide is
stable, the other is so as well. So Life in ammonia solvent is more natural
than in aqueous one.
For summarizing: Long live Aristotle! In the last 2300 years most of his answers
and explanations have been disproven; but I would
like to see the physics of 4300 AD to check the fates of our answers &
explanations. Some of the Philosopher's pictures were fundamentally good (see
Thermodynamics) and some are still not sheer lunacy (as e.g. the distinction
between Heaven and Planet in chemistry).
ACKNOWLEDGEMENT
Discussions with K. Martinás about Aristotelian and non-Aristotelian paradigms
and their relative merits in natural sciences are acknowledged. The starting
point came from these discussions, which were originally suggested by her.
REFERENCES
[1] P.
Chaunu: La civilisation de l'Europe classique. Arthaud, Paris, 1966
[2] J. N. Brönsted: Some Remarks on the
Concept of Acids and Bases. Rel. Trav.
Chim. Pays-Bas 42,
718 (1923)
[3] T. M. Lowry: The Uniqueness of Hydrogen.
[4] G. N. Lewis:
[5] J. B. S. Haldane: The Origins of
Life. New Biology 16, 12 (1954)
[6] V. A. Firsoff: An Ammonia-Based
Life. Discovery 23, 36 (1962)
[7] G.
Jander & W. Klemm: Die Chemie der wasserähnlichen
Lösungsmitteln. Springer,
[8] L.
F.Audrieth & J. Kleinberg: Non-aqueous solutions.
J. Wiley & Sons,
[9] Eva
Novotny: Introduction to Stellar Atmospheres and Interiors.
[10] K. Yanai,
H. Kojima & H. Haramura: Catalog
of the Antarctic Meteorites. NIPR,
[11] S. S. Barshay & J. S. Lewis: Chemistry of Solar Material. In: The Dusty Universe, eds. G. B. Field
& A. G. W. Cameron, Neale Watson Acad. Publ., 1975